status●

Multipele sclerose, Amyotrofische lateraalsclerose Acute kaart
Etiologie
- multicausale ziekte, exacte oorzaak onbekend
- "genetische kwetsbaarheid"
- kinderen van ouders met MS: 10 keer hogere kans
- andere helft eeneiige tweeling ook MS: 35%. Er is dus meer dan alleen genetische kwetsbaarheid.
- "genetische kwetsbaarheid"
- Factoren in puberteit en adolescentie belangrijke rol, zowel immuunsysteem als centrale zenuwstelsel in ontwikkeling[1]
- overgewicht
- (passief) roken
- tekort aan vitamine D
- In de adolescentie doorgemaakte EBV is bijna een voorwaarde is om MS te kunnen ontwikkelen.3
- Een EBV-infectie kan – in combinatie met erfelijke aanleg en de pro-inflammatoire effecten van overgewicht, roken en verlaagd vitamine D – leiden tot het ontstaan van MS. Vanuit deze gedachte is een onderzoek gestart naar EBV-vaccinaties ter preventie van MS.
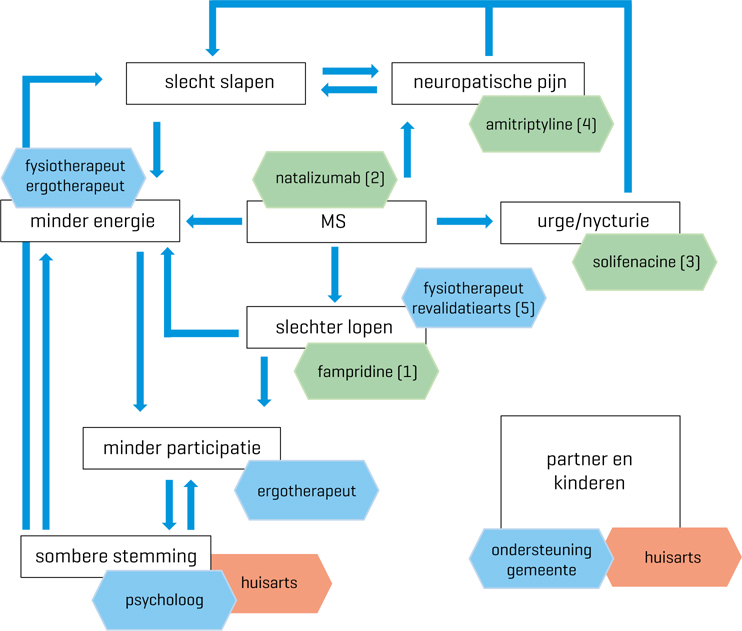
Twee vormen
- Relapsing MS
- Progressieve MS
- Oude indeling tot 2013: relapsing-remitting (RRMS), primary progressive (PPMS), secondary progressive, and progressive-relapsing phenotypes. Nieuwe indeling beter gebaseerd op realistisch beloop met dank aan verbeterende MRI-beelden.[2]
Amyotrofische Lateraalsclerose (ALS)[3]
- Neurodegeneratieve aandoening, vooral tussen het 50ste en 60ste levensjaar.
- Zeldzaam: incidentie 2-3 patiënten per 100.000 mensen per jaar
- Prevalentie is 4 tot 6 patiënten per 100.000 mensen per jaar
- Huisarts met normpraktijk ziet 2 patienten in 30 jaar
- 5-10% familiaire vorm, autosomaal-dominant
Pathofysiologie
- Deel van spectrum motorische voorhoornaandoeningen: aantasting perifeer motorisch neuron, motorische voorhoorncel (en axonen) → spierzwakte, atrofie en fasciculaties.
- Bij ALS tevens centraal motorisch neuron (piramidebaan) aangedaan → veroorzaakt vaardigheidsstoornissen en/of spasticiteit
Diagnostiek
- Klinisch beeld icm EMG,
- Na uitsluiting van andere aandoeningen bloed- en beeldvormend onderzoek
- Essentieel: minimaal drie van de vier regionen (bulbair, cervicaal, thoracaal en lumbosacraal) betrokken.
- Soms nog geen ‘clinical definitive’ ALS, maar wel duidelijk motorische voorhoornaandoening
- Bij sluipend begin soms maanden-jaar tot diagnose gesteld.
- Zowel niet onderkennen als onterechte diagnosestelling zeer belastend
Symptomen

- Vaak krachtsverlies zonder sensibiliteitstoornis in de handen.
- vaak spierkrampen
- soms benen
- soms symptomen bulbaire spieren (spraak-, kauw- en slikfunctie)
- Let op: "Handen met deuken" → Split hand[4]
- Bij vorderende ziekte
- Slikproblemen, moeizaam kauwen → gewichtsverlies
- Risico aspiratiepneumonie. Verslikken angstige ervaring, gevoel te stikken.
- Spraakstoornis, groot deel moeite verbaal uiten
- Functieverlies progressief doordat krachtsverlies toeneemt en uitbreidt naar andere delen. Toenemend ADL-afhankelijk.
- Obstipatie (verminderde mobiliteit, vochtinname en verandering van het dieet)
- Uiteindelijk altijd kortademig door spierzwakte, doodsoorzaak respiratoire zwakte al dan niet icm aspiratiepneumonie.
Voorlichting
- Website ALS-centrum (www.als-centrum.nl)
- Voor patiënten en hulpverleners
- Informatiefolder voor huisartsen en adviezen over symptomatische therapie bij verschillende klachten in verschillende fasen van de ziekte
Behandeling
- Riluzole, glutamaatremmer (enige geregistreerde therapie)
- Vertraging progressie, levensverlenging 3-6 maanden mogelijk
- Revalidatiecentrum met multidisciplinair ALS-team
- Coördinatie symptoombestrijding, ondersteuning en het aanvragen van hulpmiddelen